What Is SBS?
SBS Is a Serious and Chronic Malabsorption Disorder1
SBS is the result of physical loss and functional deficiency of portions of the intestine,
primarily due to surgical resection.1,2
The malabsorption spectrum of SBS is wide, and clinical features include1,3,4*:
Malnutrition
Dehydration
Electrolyte Disturbances
Diarrhea/Increased Outputs
*Patients with SBS require varying fluid/nutritional interventions based on individual needs.4
SBS can be a heterogeneous condition due to variations in intestinal function and remnant bowel anatomy.5
- Some patients with SBS may have intestinal failure (SBS-IF), where intestine function is below the minimum required for the absorption of macronutrients and/or water and electrolytes. For patients with SBS-IF, IV supplementation is required to maintain health and/or growth5
- Factors such as PS dependency, age, primary disease process, and comorbidities can affect the overall prognosis in patients—including survival rates and life expectancy, which can vary1
Patients with SBS-IF are unable to increase their oral intake sufficiently or absorb enough nutrients. They often require PS.5
SBS Can Have a Functional or Anatomical Origin1,2
An SBS diagnosis can involve identifying one or more possible pathophysiologies, underlying diseases, or congenital conditions. As a heterogeneous condition, SBS can result from a number of etiologies or factors.1,2
Common causes of SBS6-11:
This is not a complete list of causes for SBS.
| Adult Patients | Pediatric Patients |
|---|---|
| Vascular events (eg, embolism, thrombosis) | Necrotizing enterocolitis |
| Crohn's disease | Gastroschisis |
| Complications from bariatric surgery | Midgut volvulus |
| Malignancy | Malrotation |
| Trauma | Intestinal atresia |
| Volvulus | Intestinal ganglionitis |
| Strangulated hernia | Trauma |
| Small bowel fistulas | Hirschsprung's disease |
| Ulcerative colitis | Crohn's disease |
Patients with SBS have reduced intestinal absorption12,13
Patients with SBS may have limited GLP-2 secretion due to the removal of L cells following resections12,13
Any resection can impact GLP-2 secretion resulting in SBS.12,13
No Colon-in-continuity
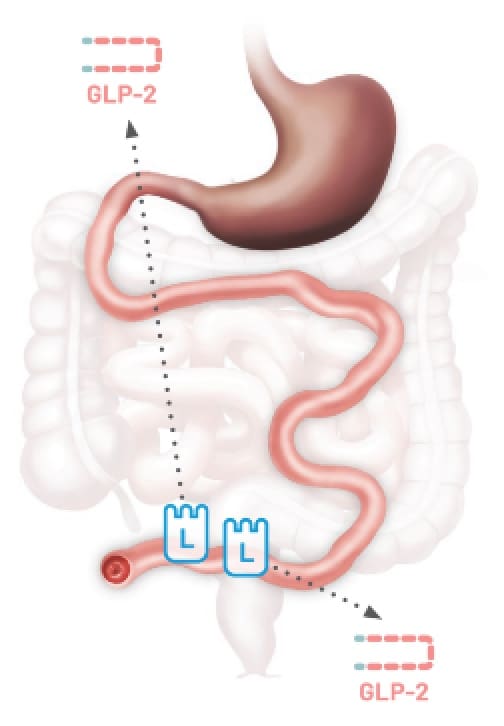
Type 1
End
Jejunostomy14,15
Colon-in-continuity
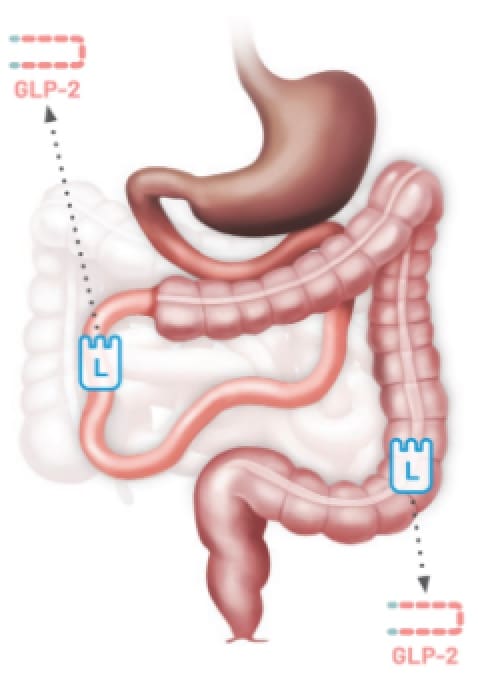
Type 2
Jejunocolonic
Anastomosis14,15
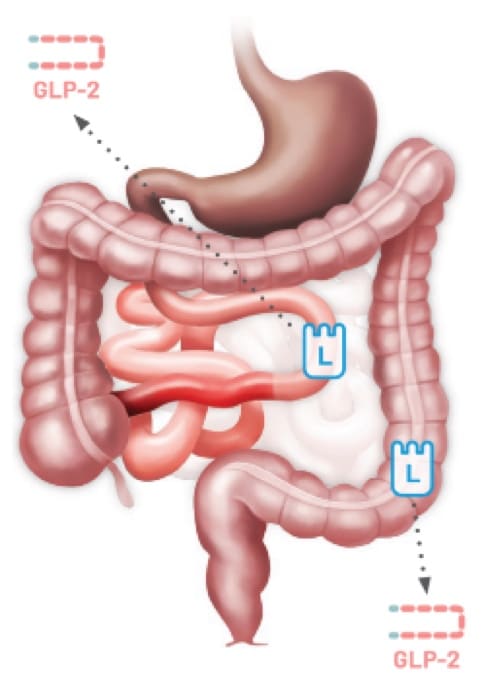
Type 3
Jejunoileal
Anastomosis14,15
This is not a comprehensive list of bowel resections.
Images are for illustrative purposes only. GLP-2 secretion after resection will vary, and different anatomies may lead to SBS.
Patients with SBS may go undiagnosed due to underreporting and the lack of reliable patient databases.9
Diagnosis codes
ICD-10-CM codes for short bowel syndrome16
K90.82 Short bowel syndrome
K90.821 Short bowel syndrome with colon-in-continuity
K90.822 Short bowel syndrome without colon-in-continuity
K90.829 Short bowel syndrome, unspecified
Management of SBS is complex and requires an individualized approach17
Important treatment goals for patients with SBS17,18
While there is no cure for SBS, a multidisciplinary team can help manage it. Experts can include physicians, surgeons, nurses, dietitians, and/or social workers who may contribute to the success of achieving patients' treatment goals.17,18
- Maintain adequate nutrition and hydration requirements7,19
- Reduce or eliminate the need for parenteral support (PS) and increase oral and/or enteral feeding7,19
- Promote intestinal adaptation7,19
- Minimize disease- and parenteral support–related complications7,19
Patients with SBS often require parenteral support (PS)19
PS dependency can vary in nutritional components, frequency, and administration times.
Less nutrients
Intravenous (IV) Fluids20
Delivers hydration along with electrolytes and potentially other components (ie, dextrose 5% and 0.45%, normal saline, lactated Ringer’s).
More nutrients
Parenteral Nutrition21
Provides balanced caloric energy needs and can include a variety of components, such as protein, fat, and sugar, as well as electrolytes and vitamins.
Peripheral parenteral nutrition (PPN) Delivered through a peripheral line
Total parenteral nutrition (TPN) Delivered through a central venous line
PS is lifesaving for patients with SBS but does not increase intestinal absorption4,19
Patients often require PS to make up for reduced absorption. PS is supplementary nutrition and does not increase intestinal absorption, which will continue to be an issue for patients with SBS.4,19
Patients with SBS often require PS, but long-term use of PS is associated
with numerous complications, such as5,7,10,22†:
Hepatobiliary
Diseases ie, intestinal failure–associated liver disease [IFALD] and gallstones
Kidney
Diseases ie, hyperoxaluria
Central Venous
Complications ie, septic infections, thrombosis, and loss of vascular access
Metabolic
Bone Disease ie, osteoporosis
A reduction in PS requirements may give patients more freedom for work, hobbies, school, sleep, rest, and social interaction.10,23‡
†The effects of GATTEX on these complications were not studied.
‡Examples of how some patients may spend their time if they are able to achieve a reduction in PS requirements are for illustrative purposes only. Patients should always discuss their individual medical circumstances and activities with their doctor.
Glucagon-like peptide-2 (GLP-2) plays an important role in intestinal absorption14,24
GLP-2 is an intestinal hormone that maintains the structure and function of the intestine to facilitate absorption. Patients with SBS may have limited GLP-2 secretion due to the removal of L cells following resections.14,24
Increase Villus Height
& Crypt Depth13§
Facilitate Absorption
of Nutrients13,25-28§
Increase Intestinal
& Portal Blood Flow24,26,29,30§
Inhibit Gastric
Acid Secretion24,26,29,30§
§Based on studies in animals and healthy volunteers; the clinical relevance of these data has not been established.